Qarad - European Regulatory Services
For a long time, paper Instructions For Use (IFU) have been the norm in the medical device industry. However, many manufacturers are now investigating the possibility of replacing the paper IFU with electronic Instructions For Use (eIFU), provided to the user online. Such a web-based solution has many advantages, but presents challenges in complying with all regulatory requirements and dealing with its major impact on internal processes. Outsourcing the project to an experienced partner can greatly reduce the efforts for the implementation of an eIFU solution.
The advantages of eIFU cannot be underestimated. By replacing the paper IFU with their digital versions, manufacturers significantly reduce their economic and environmental costs, while customers enjoy an improved customer-experience.
Traditionally, paper IFU are known for their small fonts and unattractive appearance as a consequence of fitting a lot of information in many languages on the smallest possible paper or booklet. Though everyone got used to this unattractive, non-user-friendly and hardly legible format, providing users with access to IFU online to print once or to consult on smartphones, tablets, computers or intranet would significantly enhance the user experience. The limitations on font and lay-out do not apply to the digital format. Digitizing documentation offers room for improving design and layout and allows users to only select those languages of interest to them, eliminating the need to browse through pages upon pages of information in a language not their own.
From an operational perspective, the electronic distribution of IFU has many merits. If IFU are no longer printed, the industry saves money on large amounts of paper and on printing. Removing the paper from the boxes will also allow companies to reduce the box sizes, thus saving money on product packaging and increasing the storage capacity of the warehouse. Moreover, leaving out paper IFU from the box impacts the packaging process. It eliminates the need for having the right IFU at the right time, eliminates the step of adding the IFU to the box and eliminates the errors that may occur in those steps. It also makes the in-process controls to verify whether the right document went into the right box obsolete. Furthermore, in case of IFU related nonconformities, the manufacturer is no longer faced with the physical replacement of the paper IFU in the packages.
The concern for patient risk may be a reason companies are not implementing eIFU yet. How can patient safety be ensured when clear working instructions are not provided with the product? Contrary to the expectations, for many medical devices (MD) and in vitro diagnostics (IVD), eIFU actually have characteristics that help to improve patient safety. The improved legibility of the IFU and the absence of constraints on the volume of information provided, reduce the occurrence of user error. In a professional environment, a clear indication how to obtain the eIFU enables users to access the right document at any time. Moreover, the electronic version allows regular, instant updates to the instructions and it eliminates the risk of a paper document getting lost once the package is open.
These numerous advantages are creating a shift towards eIFU to become the future norm in the MD and IVD industry. Nevertheless, current regulations still limit the use of eIFU to all medical devices.
The first objective of regulations is to ensure the safety of patients, users and other persons. Therefore, the electronic distribution of IFU is the subject of European regulations and guidance and is also regulated in other territories.
Patient safety can only be guaranteed if eIFU are used in the right setting and under appropriate conditions. It is important to understand the regulatory framework in which eIFU can be used. In this article, the focus is put on the European eIFU regulations.
First of all, replacing the paper instructions by eIFU is not allowed for all MD and IVD. eIFU are only accepted for devices to be used by a healthcare professional. All devices intended for lay people require a paper IFU to be supplied together with the product. For IVD used by healthcare professionals, eIFU can be provided instead of paper IFU, except for point-of-care devices. The regulators fear that the user in a point-of-care setting might not have easy access to the eIFU at the time of use of the IVD. Furthermore, special rules also apply for IVD instruments and software.
For MD, the regulations are even more restrictive. There are five clearly defined types of MD for which IFU can be made available online (see table below). All others still require a paper copy to accompany the device.
Only for professional use!
Second, even with an electronic system in place to provide eIFU, some users may still want a paper copy of the IFU. Therefore, a compliant eIFU solution must also provide the users with a way to obtain a paper copy upon request. For IVDs, the manufacturer must provide a free telephone number which the user can call to request the paper IFU to be sent to him via mail, fax or email. For medical devices, the regulation does not specify how a copy must be provided. It does clearly state, though, that the paper version must be provided to the user within 7 days from the request, and this without any charges to the user.
Third, uploading documents to a digital platform may appear quite straightforward, but the platform itself must comply with certain requirements as well. From a functional perspective, the website must work properly. Retrieving an IFU from the website should be self-explanatory in a language easily understood by the end-user. Access to the IFU should be quick and easy. The content of the instructions from the eIFU website, should be identical to the paper IFU and in a read-only format such as pdf. Moreover, the system must be secured and as such be hosted in an environment protected against hardware and software intrusion. This is also important to ensure permanent access to the website.
From a regulatory perspective, more restrictions apply to an eIFU platform. For instance, privacy regulations do not allow to capture personal data of the users downloading eIFU without their consent. However, regulations do not only restrict the use of eIFU. The new European MD and IVD Regulations clearly recognize the increasing importance of eIFU and state that the manufacturer has to make IFU available and keep them up to date on their website. This applies to all the devices, but except for the product groups described above, should be done in addition to the paper IFU accompanying the product. Manufacturers could consider meeting this requirement as an opportunity and first step to remove paper when possible, provided that they immediately use a web platform that meets the regulatory requirements for eIFU.
The decision to shift towards eIFU should not be made lightly. It has a significant impact on the manufacturer’s quality management system and will have to meet Notified Bodies’ expectations during audits.
A switch to the electronic distribution of IFU influences many quality system processes. The web platform itself will require full validation and the risk for patients and users has to be assessed. This risk assessment needs to focus on the risks associated with eIFU compared to paper versions, taking the use of the product, the environment of use and other end-user’s needs into consideration.
The implementation of an eIFU solution will have an impact on procedures and instructions in the management of eIFU and label content, the inventory management of labeling, the packaging process, logistical process, the ERP system and will trigger the creation of new procedures for the management of the web platform itself. Although this will initially take some effort, it will considerably reduce the work load of these processes. It will for example facilitate the process of introducing a new version of the IFU, since it will a.o. not impact the destruction and replacement of obsolete inventory. Paper IFU would require to be reprinted and if it concerns an immediate correction of an IFU boxes in stock must be reopened and IFU replaced. This is very labor-intensive compared to the electronic alternative.
Notified Bodies will verify whether the eIFU solution has been properly validated, whether risk assessment has been done, quality system processes and operational instructions have been adapted and labelling modified.
Manufacturers that use eIFU must inform the user on how to obtain the instructions for their products. It is recommended to do so via the product’s labeling.
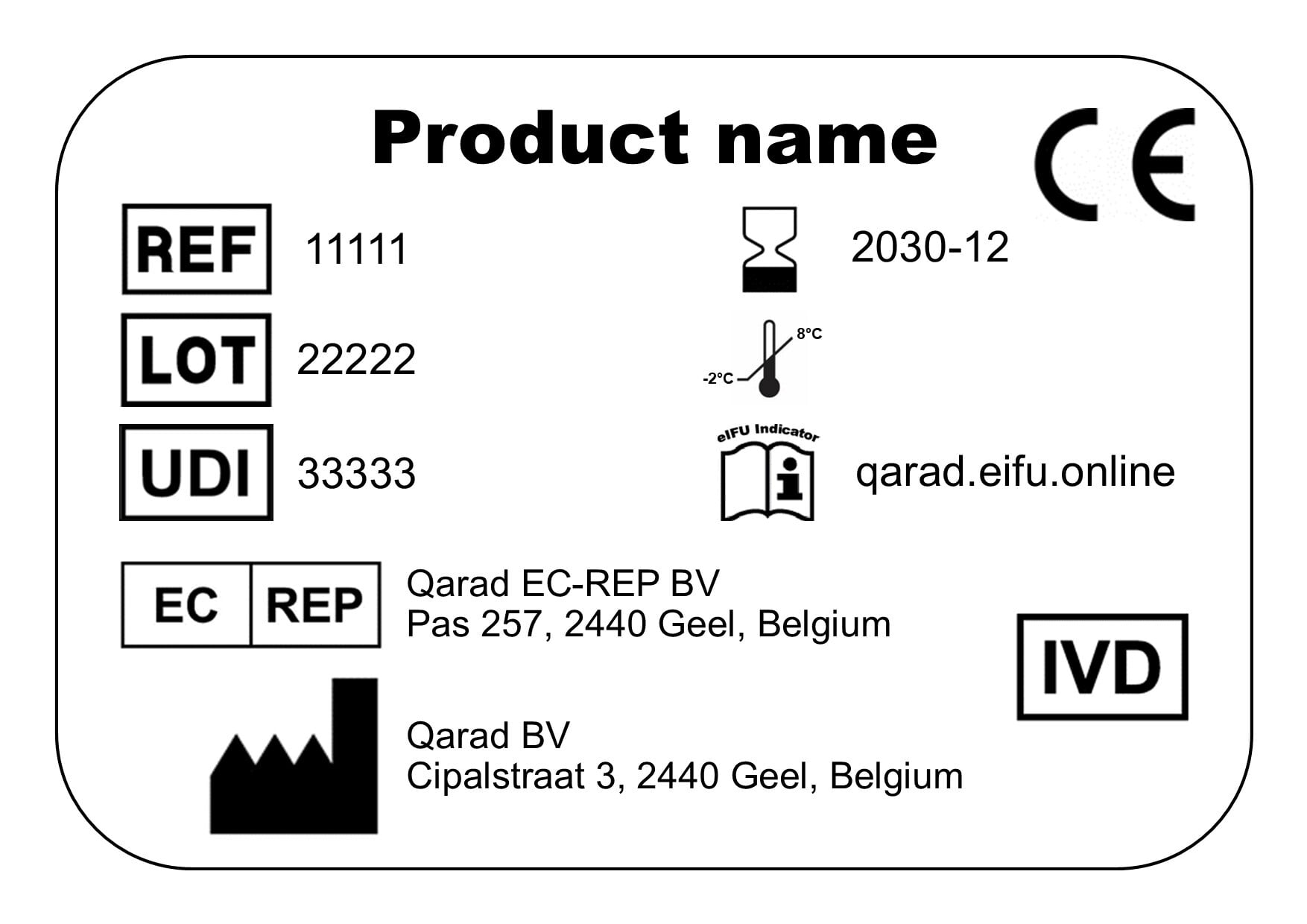
Clear instructions must be provided on how to obtain the respective IFU, including a unique reference to identify the correct version of the document. This identifier has to be defined by the manufacturer and could be the reference number, UDI, GTIN or any other unique identification assigned to the product. As mentioned before, whenever the IFU have been revised, a clear indication must also be added to alert the customer that the document has been changed. Last but not least, a customer must be provided with the option to obtain the IFU on paper upon request as alternative for the eIFU. The necessary information on how to request that paper copy should be included on the label as well, or communicated by other means.
It clearly takes a significant effort to implement an electronic system for the distribution of IFU, that complies with the different regulatory, functional and operational requirements. However, the investment will quickly create a return by savings in direct costs and by increasing the efficacy of the processes related to IFU management. The implementation of an eIFU solution becomes considerably less burdensome by working with a reliable supplier, who not only offers a ready-to-use, fully compliant eIFU solution, but who also understands the full context of an implementation project and provides the manufacturer the necessary support in e.g. validation, risk assessment, labeling etc… With such a partner, the manufacturer is not alone and what first looked impossible, suddenly becomes a solution within reach.
Qarad is the pioneer in eIFU and is still global leader. Its eIFU solution complies with all regulatory requirements and has all the functionalities that manufacturers expect built-in. Qarad assists its customers by providing risk assessment and software validation documentation, by giving recommendations in handling labeling changes, notified body change requests, etc… Qarad not only provides a platform, but also facilitates its implementation, reducing the manufacturer’s resource needs.