Analytical and preparative chromatography in mRNA production
Rok Sekirnik, Head Process Development mRNA | pDNA, Sartorius BIA Separations, d.o.o.
The use of mRNA vaccines in the fight against SARS-CoV-2 during the COVID-19 pandemic, has demonstrated their enormous potential as a highly effective therapeutic modality and has led to a revolution in biomedicine. Since then, mRNA has also shown promise in the treatment of cancer, rare diseases, anaphylaxis, CRISPR-Cas9-based therapy and others. There are currently >100 clinical trials registered with mRNA being the drug substance. The technology to produce mRNA therapeutics can still be considered as being in the early stages of development, as multiple academic and industrial laboratories across the world invest significant funding and talent, with the intention to increase productivity and purity in the production of mRNA drug substance and lipid nanoparticles that would deliver affordable mRNA therapeutics. This article aims to present a technological paradigm of harnessing the potential of chromatography as a purification tool with high selectivity and scalability that are unmatched by other purification methods, as well as analytical technology that can provide timely and accurate information on the quality and quantity of the mRNA product, its precursors and side-products.
mRNA is produced by an in vitro transcription (IVT) reaction. It is a multi-component enzyme-catalysed reaction that uses a DNA template for mRNA production. The DNA template is specific for each mRNA construct, so the process of mRNA production thus really begins with plasmid production (Figure 1).
Plasmid DNA (pDNA) is produced through a fermentation process in Escherichia coli, a gram-negative bacterium that has been widely used in microbial research and in the production of various biopharmaceuticals. After bacterial fermentation and cell harvest (with centrifugation or tangential flow filtration), the target pDNA is extracted from the harvested cell paste and purified to remove host cell proteins, RNA, genomic DNA, and other impurities. Alkaline lysis, which has remained largely unchanged since the 1970s [1], is used to lyse the bacterial cells to extract pDNA. Scaling up the lysis process can be challenging due to the need for precise control over the duration of time cells are exposed to a strong base (NaOH), which is difficult to achieve in a large batch process. Prolonged exposure of bacterial cells to a strong base and to high shear forces (due to impeller motion required for mixing) can lead to fragmentation of host-cell genomic DNA, as well as shearing/nicking of the supercoiled (SC) plasmid, resulting in the open-circular (OC) DNA. Both are critical impurities that need to be removed in the downstream process. In-line/continuous lysis has the advantage of having precise control over cell exposure to NaOH during cell lysis with flow adjustment and efficient neutralization of lysed cells by an inline static mixer. This improves robustness and contaminant profile (genomic DNA, isoform homogeneity) [2].
To monitor the efficiency of alkaline lysis, rapid at-line HPLC analytics is implemented in the downstream process. Anion exchange (AEX) HPLC method utilizing CIMac™ pDNA analytical monolith quantifies the main pDNA isoforms (SC, OC, linear - LIN, and multimer) as well as detects the presence of RNA and other impurities in the sample [3]. This is possible from the point of lysis, where the concentration of pDNA is still very low (approximately 0.02 mg/mL) and not easily analysed with other analytical methods which are either not quantifiable (AGE) or would require a prohibitive volume of sample to exceed the limit of quantification (CE). Due to the physical properties of monolith columns, the volume of analyte that can be applied onto the column is in principle unlimited (limitation is only the binding capacity for the target analyte); very low concentrations of analyte can thus be analyzed simply by applying a larger sample volume.
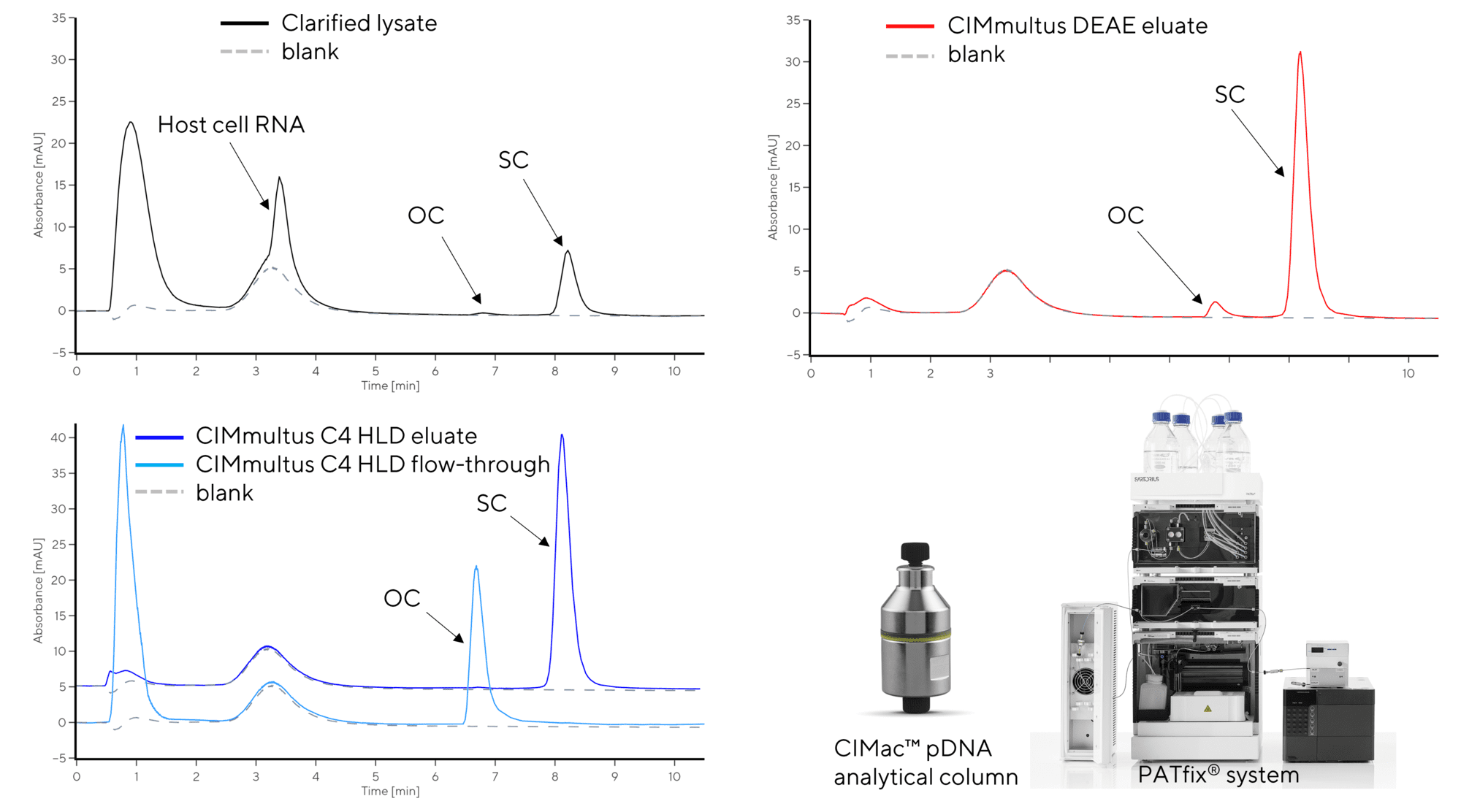
Parameters that are commonly studied during optimization of alkaline lysis process are cell resuspension ratio (5 – 20x), concentration of sodium hydroxide (NaOH) and sodium dodecyl sulfate (SDS) in the lysis buffer, lysis contact time (2-10 min), concentration and pH of the neutralizing buffer, duration of neutralization, concentration of flocculating agent (e.g. CaCl2) and duration of flocculation. Due to a complex interplay of factors, design-of-experiment (DOE) approach using a suitable software, e.g. MODDE®, is typically used to derive the design space. Analytical AEX monolith provides flow-rate independent (i.e. rapid) resolution and quantification of sample components – unbound impurities (e.g. proteins), weakly bound host-cell RNA and plasmid isoforms. This provides insight both into the efficiency of lysis (mass of plasmid extracted per mass of cell paste, relative to the mass of RNA) and quality of the pDNA extracted (e.g. degree of open-coil isoform generated during lysis, Figure 2).
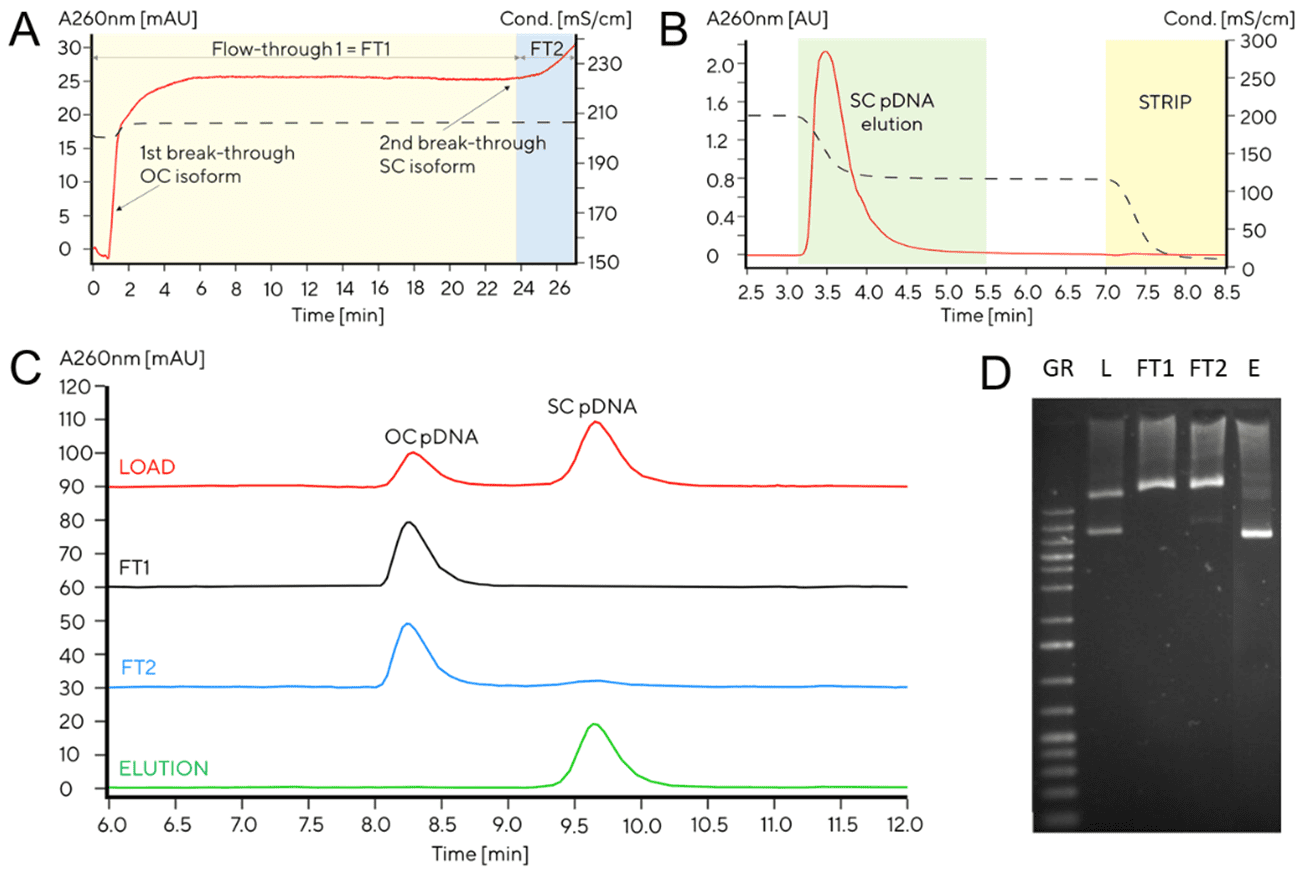
Depending on the end application for the selected plasmid, a polishing step using hydrophobic interaction chromatography (HIC) via CIMmultus® C4 HLD may be necessary. This step removes unwanted plasmid isoforms, e.g. OC pDNA and residual genomic DNA. This results in a high SC pDNA purity (>95%), which is required if the plasmid is to be used as a naked vaccine or a transfection agent. Another function of C4 HLD is that it also achieves a high clearance of proteins and endotoxins in order to meet regulatory specifications for plasmid DNA vaccines. Removal of pDNA isoforms can be monitored using the CIMac pDNA analytical column (Figure 3).
Plasmid linearization
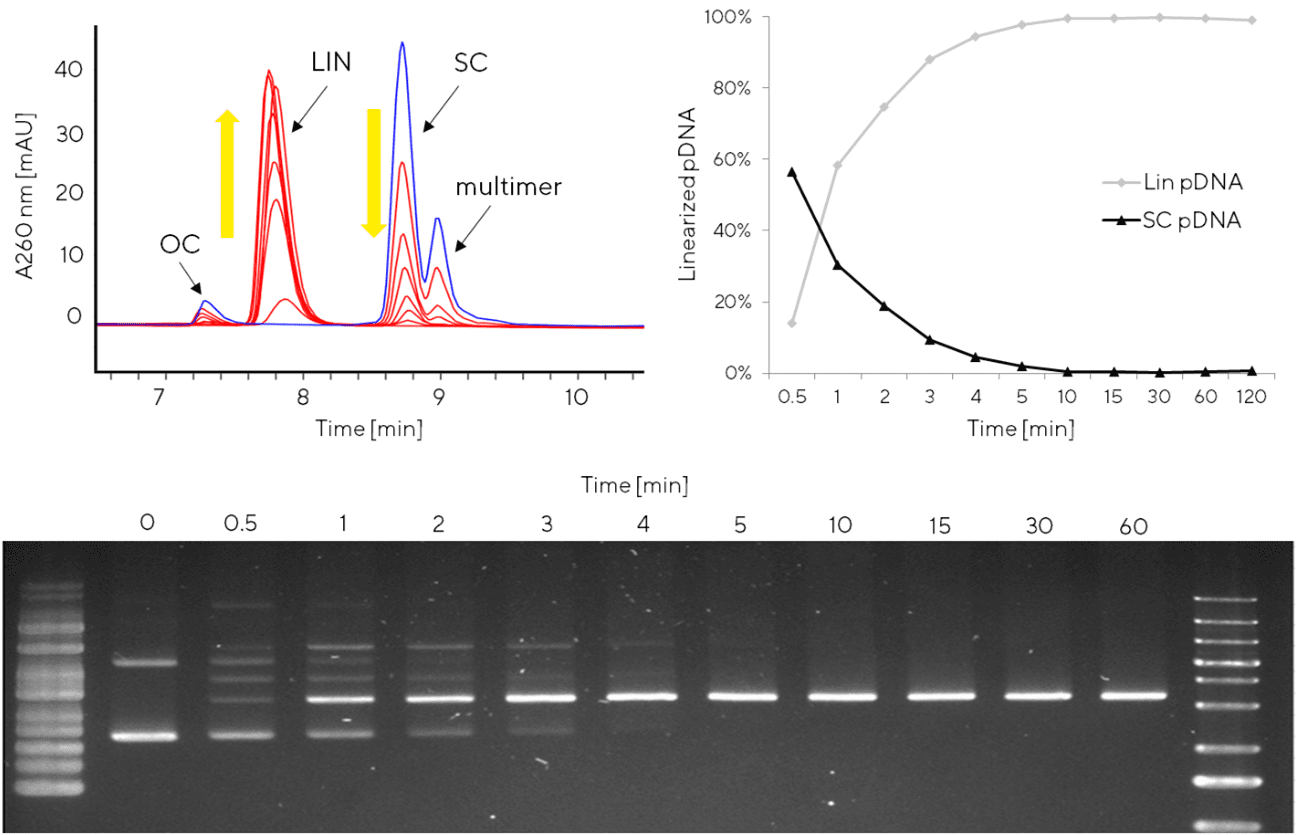
pDNA is linearized using a suitable restriction enzyme, selection of which is construct-dependent (widely used enzymes are NotI, BspQI, NdeI). The linearization protocol can be optimized for kinetics and yield by adjusting incubation temperature, restriction enzyme concentration and other factors. Quantifiable data of linearized product versus starting circular material is desirable for optimal interpretation of study results. AEX HPLC, e.g. the CIMac pDNA analytical method described above for plasmid purification can also be used to monitor plasmid linearization without any modification. Compared to traditional semi-quantitative approaches to monitor restriction enzyme activity (e.g. agarose gel electrophoresis), HPLC allows area-under-curve to be integrated for both circular and linearized isoforms (Figure 4). A robust analytical method is useful when validating the linearization process (e.g. comparing multiple enzyme batches), undergoing technology transfer or performing scale-up studies.
After linearization, the product contains the desired linearized pDNA, as well as the restriction enzyme, cofactors and other residual host cell contaminants, if they were not removed during plasmid purification process (e.g. endotoxins). Using HIC chromatography operated in bind-elute mode, which is generic with respect to plasmid size, the product of linearization can be purified and prepared for the next step in mRNA production. Under HIC conditions, proteins and endotoxins bind more strongly than plasmid, which is eluted in a defined step of salt concentration. The salt (ammonium sulfate) is removed with TFF using a membrane cut-off size of 10-30 kDa. This is counterintuitive because of the large size of the plasmid (>1 MDa), hinting at interesting biophysical properties of the plasmid after linearization (spaghetti-sieve mechanism), which have not yet received due academic attention.
IVT reaction
mRNA is produced by in vitro transcription (IVT) reaction, a multi-component enzyme-catalysed reaction using RNA polymerase for mRNA production from a DNA template. At its core, the IVT reaction requires a linear double-stranded DNA template (linearised pDNA), RNA polymerase and NTPs (A, U, C, G as wild-type or modified, e.g. N1-methylpseudouridine) as building blocks to create new strands of mRNA and Mg2+ as a co-factor required for incorporation of NTPs [9]. The catalytic efficiency of RNA polymerase can be increased by optimizing the reaction conditions (pH, temperature) as well as the concentration of NTPs, Mg2+, DNA template and additives that increase the solubility of RNA (spermidine) or the activity of polymerase (DTT).
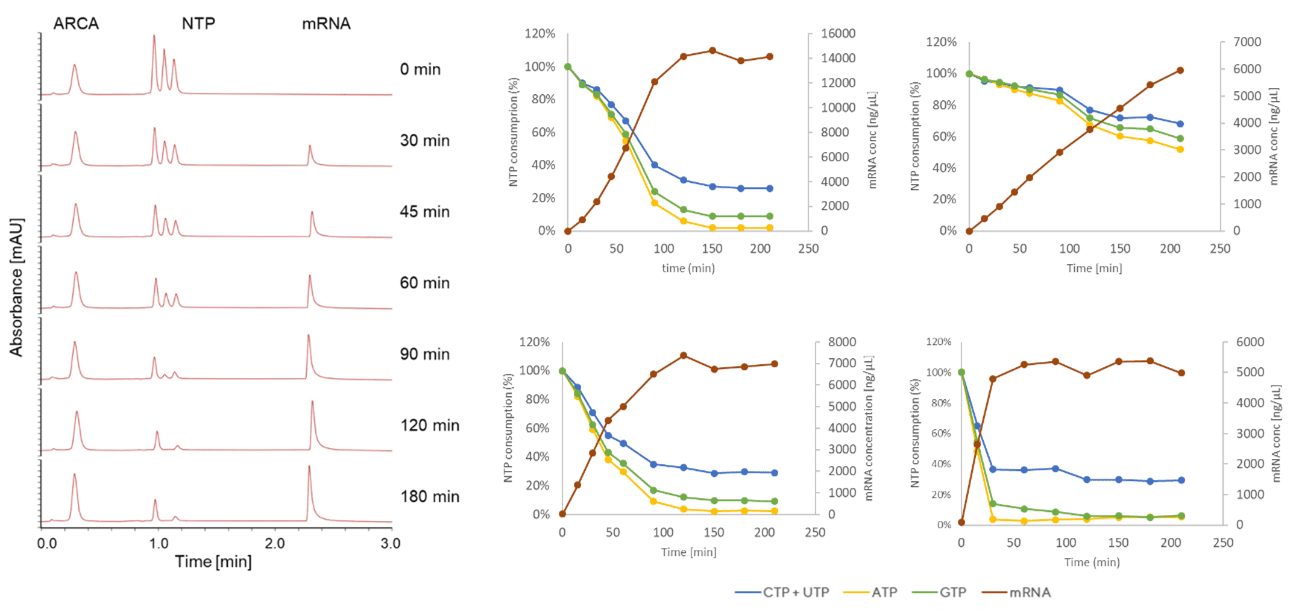
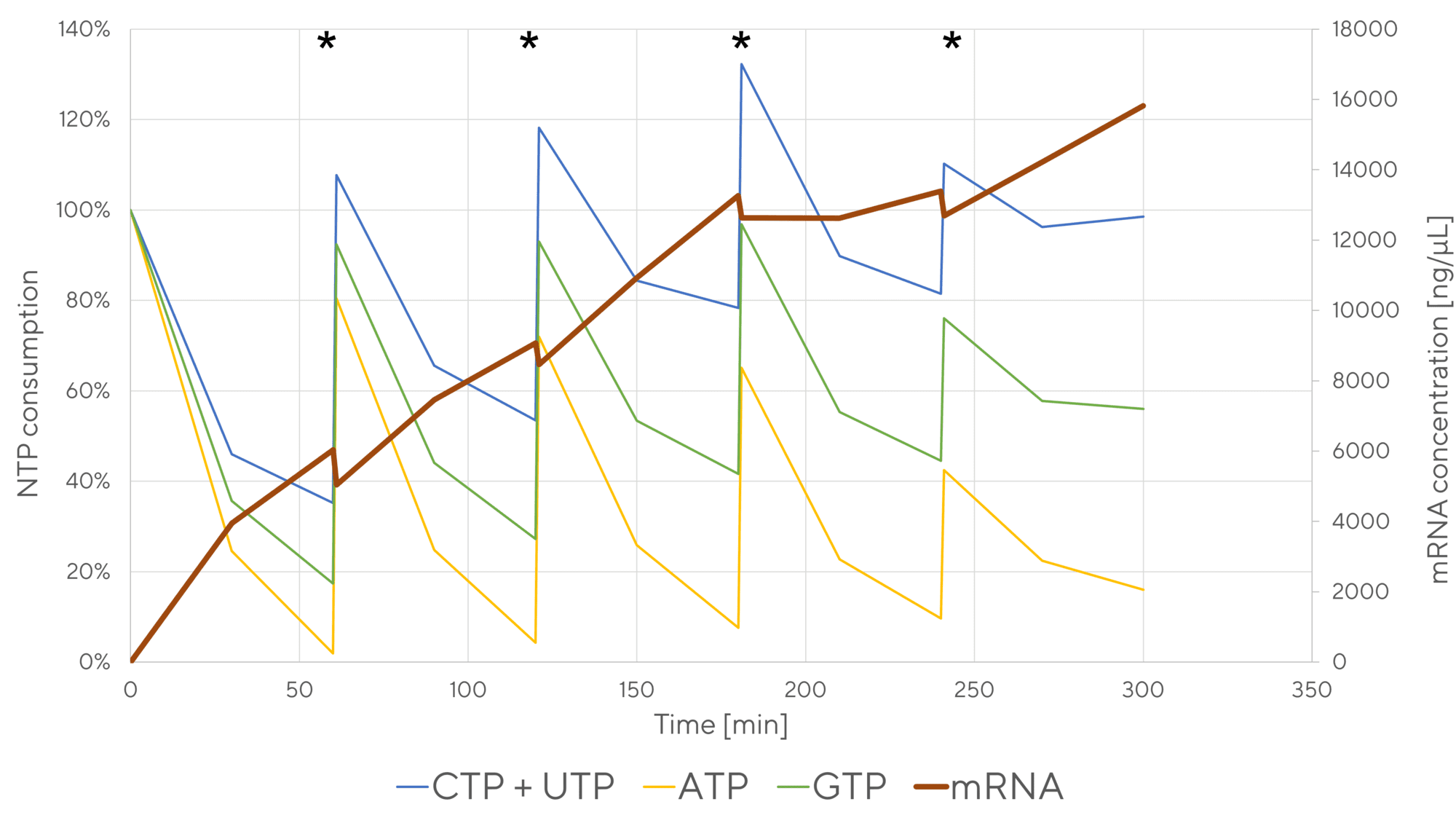
The IVT reaction can be monitored by HPLC to determine the concentrations of IVT components as a function of time. CIMac PrimaS™ monolithic column was developed to monitor levels of capping reagent, NTPs, pDNA and mRNA with a resolution time of 3 min (Figure 5), allowing monitoring of mRNA production kinetics with a minimal analytical lag [10]. Unlike traditional single-attribute analytical approaches that rely on fluorescent dye (e.g. Ribogreen) binding to nascent mRNA, HPLC allows us to study the effects of specific reaction conditions on the consumption of NTPs and capping reagent in regards to the production of mRNA. Kinetic data of NTP consumption/mRNA production is important for understanding polymerase kinetics, lot-to-lot comparisons of IVT reagents (particularly polymerase, which is prone to oxidation), reaction scale-up and transferring the process to a different manufacturing or development site. Coupled with DOE study design, a design space can be derived supporting the development of mRNA therapeutics according to QbD principles. A generic analytical method applies to the production of nucleic acid therapeutics ranging from tRNA (70 nucleotides), mRNA (1000 – 5000 nucleotides) to saRNA (10,000 – 12,000 nucleotides) and circular RNA.
In principle, it is possible to convert the batch IVT reaction to a fed-batch reaction (addition of reagents - NTPs and Mg2+ during the reaction), due to its catalytic nature [11]. To avoid substrate inhibition or reaction pausing, on-line or at-line monitoring of reagent concentration is required. Currently, only HPLC can provide sufficient resolution of NTPs, capping reagent, pDNA and mRNA, and monolith columns are able to resolve the reagents with a resolution time of 3 min, offering suitable temporal resolution in the context of reaction time (2-3 hr). NTPs (in complex with Mg2+) are added to the IVT reaction at time-points identified by CIMac PrimaS HPLC analytics to prevent depletion and over-accumulation of NTP/Mg2+, which could stall the reaction by inhibiting the polymerase and potentially hydrolytically cleave mRNA.
Bolus feeding can be further developed into continuous feeding (Figure 6). This requires a (bio)reactor with continuous feed addition capability, such as AMBR250, BioStat RM or other bioreactors commonly used in biopharmaceutical production. The rate of feed addition is determined based on kinetics of NTP depletion studied in bolus fed-batch IVT reactions and translated to continuous feed kinetics. Applying this approach, over 2 g of mRNA was produced from a starting volume of 100 mL in an AMBR250 benchtop bioreactor [12].
mRNA Purification
Various mRNA purification strategies have been proposed in the literature, which reflects both the creativity of process developers and differences in the production strategy of mRNA itself (Figure 7). Four main strategies of IVT are used, in order of decreasing popularity: i) co-transcriptional capping with co-transcriptional polyadenylation, ii) post-transcriptional capping with co-transcriptional polyadenylation, iii) co-transcriptional capping with post-transcriptional polyadenylation and iv) post-transcriptional capping with post-transcriptional polyadenylation [13]. The most common IVT approaches (co-capping/co-tailing and post-capping/co-tailing) lend themselves to affinity purification by poly-deoxythimidynilic acid ligand (oligo-dT) immobilized onto a chromatographic support (porous particle or monolith), which removes 99% of all IVT impurities in a well-defined, scalable purification [14]. Oligo-dT may be preceded by a TFF step (buffer-exchange, removal of low-molecular weight impurities), which does not increase purity on its own compared to Oligo-dT. IVT reaction product is adjusted to Oligo-dT loading conditions by dilution in the salt-containing buffer. An in-line dilution approach is often applied to minimize the hold-time of mRNA in high salt conditions.
To our surprise, mRNA (eGFP) purified with Oligo-dT column was stable for several weeks at room temperature and even at 37°C, in contrast with mRNA purified with commercial isolation kits, which rapidly degraded [15]. Oligo-dT resin exists in 96-well plate format allowing for fast and robust screening of binding and elution conditions.
Post-transcriptional capping strategy requires an additional purification step after mRNA is captured from IVT and capped (e.g. with Oligo dT), to remove capping machinery (capping enzyme and reagents). This can be performed with another affinity step, or a polishing step that removes the capping machinery together with product-related impurities (e.g. dsRNA, see below).
Post-transcriptional polyadenylation precludes the use of affinity chromatography. Instead, TFF, precipitation or multimodal chromatography (or in very rare examples -HIC) can be applied to capture mRNA from IVT reaction. Multimodal column PrimaS combines properties of AEX and hydrogen bonding. In contrast to the Oligo-dT process, it uses a pH gradient in combination with a high salt wash to purify the mRNA sample. We have developed a method of instant neutralization of pH after elution. Purified mRNA is stable at room temperature and even at 37°C [Megušar 2023, in press]. A similar approach can be applied to circular RNA.
Chromatography provides selectivity coupled with online monitoring that other purification approaches cannot meet – operating PrimaS multimodal column on a chromatography skid equipped with UV detectors provides real-time information on the separation of proteins and DNA from RNA; if a chromatography skid is equipped with fluorescence detection, the sensitivity of protein clearance can be increased at least by an order of magnitude.
mRNA polishing
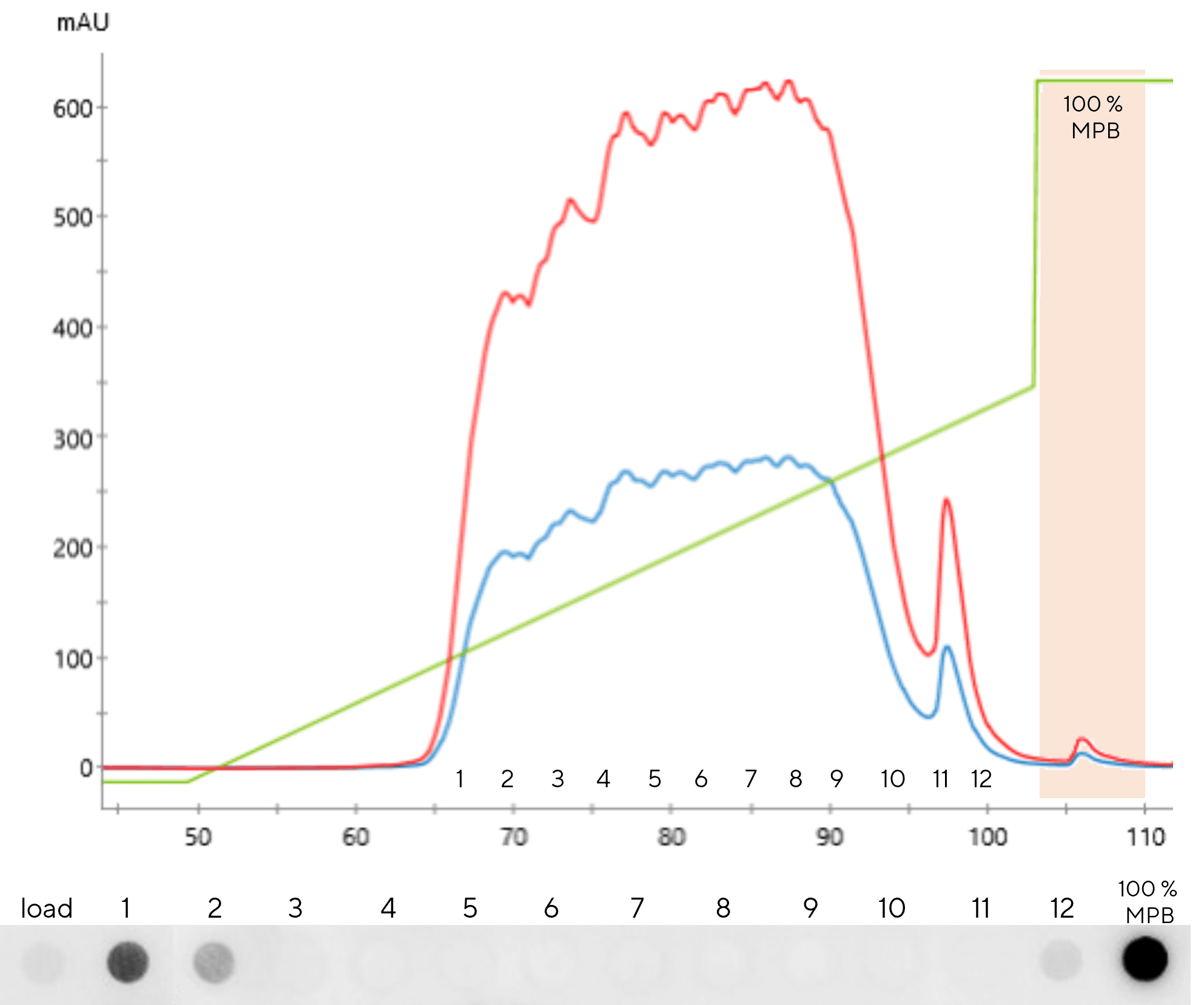
The most commonly used polishing approach for mRNA is reverse-phase chromatography [16]. In monolith format, the most suitable column chemistry of reverse-phase purification is styrene divinyl benzene (SDVB), which combines the benefits of flow-rate independent chromatographic properties (resolution, capacity) with the advantages of reverse-phase chromatography for mRNA polishing to remove undesired dsRNA contaminants from single-stranded mRNA. Purification utilizes denaturing conditions (temperature, organic solvent) to chemically separate the RNA chains forming double-stranded moieties, and chromatographic separation by hydrophobicity to separate the denatured strands by size. Binding of highly negatively charged mRNA to hydrophobic resin is enhanced with ion pairing reagents, such as triethylammonium acetate. Short fragments elute early in the acetonitrile gradient, 3’ extended dsRNA products elute late in the gradient and the target single stranded RNA elutes in the middle of the gradient. This method is scalable to a 8 L CIM SDVB column, enabling the purification of ~ 10 g of mRNA per batch.
A similar chromatographic set-up can be applied in an analytical setting to monitor the purity of DSP product – analytical reverse-phase monolith (CIMac SDVB) operated in acetonitrile gradient will separate RNA molecules by size (Figure 8).
Conclusions
In order to build quality into the mRNA production process, development should be supported with at-line analytics. Chromatographic analytical methods, which are fast and able to deconvolute multiple quality attributes at a time can support optimisation throughout the entire process of mRNA production starting with plasmid isolation. Two generic analytical methods can, without optimization, support the majority of development activities related to plasmid and mRNA production. CIMac pDNA analytical column offers quantification of pDNA isoforms from alkaline lysis of E. coli cell paste through to sterile filtration of SC pDNA. CIMac PrimaS analytical column is used to monitor IVT reaction components and quantify mRNA from the early stages of IVT to the final polishing steps.
Author would like to thank Ajda Sedlar, Klemen Božič, Tjaša Valič, Kevin Mozetič and Kaja Tudja for help during preparation of the manuscript.
References
[1] Birnboim, H. C., Doly, J. Nucleic Acids Research 1979, 7, 1513–1523.[2] Urthaler, J., Ascher, C., Wöhrer, H., Necina, R. Journal of Biotechnology 2007, 128, 132–149.[3] Pavlin, N., Černigoj, U., Bavčar, M., Plesničar, T., Mavri, J., Zidar, M., Bone, M., Kralj Savič, U., Sever, T., Štrancar, A. ELECTROPHORESIS n/a.[4] Černigoj, U., Štrancar, A. Methods Mol Biol 2021, 2197, 167–192.[5] Cardoso, S., Černigoj, U., Lendero Krajnc, N., Štrancar, A. Separation and Purification Technology 2015, 147, 139–146.[6] Kralj, Š., Kodermac, Š. M., Bergoč, I., Kostelec, T., Podgornik, A., Štrancar, A., Černigoj, U. ELECTROPHORESIS n/a.[7] Krajnc, N. L., Smrekar, F., Cerne, J., Raspor, P., Modic, M., Krgovic, D., Strancar, A., Podgornik, A. J Sep Sci 2009, 32, 2682–2690.[8] Krajnc, N. L., Smrekar, F., Štrancar, A. Journal of Chromatography A 2011, 1218, 2413–2424.[9] Milligan, J. F., Groebe, D. R., Witherell, G. W., Uhlenbeck, O. C. Nucleic Acids Res 1987, 15, 8783–8798.[10] Kostelec, T., Sekirnik, R., Martinčič Celjar, A., Nemec, K. Š., Gramc Livk, A., Gagnon, P., Štrancar, A. Eliminating the Analytical Bottleneck in Production and Purification of mRNA. BioProcess International 2021.[11] Pregeljc, D., Skok, J., Vodopivec, T., Mencin, N., Krušič, A., Ličen, J., Nemec, K. Š., Štrancar, A., Sekirnik, R. Biotechnology and Bioengineering 2023, 120, 737–747.[12] Skok, J., Megušar, P., Vodopivec, T., Pregeljc, D., Mencin, N., Korenč, M., Krušič, A., Celjar, A. M., Pavlin, , Sekirnik, R. Chemie Ingenieur Technik 2022, 94, 1928–1935.[13] Rosa, S. S., Prazeres, D. M. F., Azevedo, , M. P. C. Vaccine 2021, 39, 2190–2200.[14] Mencin, N., Štepec, D., Margon, J., Dolenc, D., Simčič, T., Rotar,, A., Černigoj, U. Separation and Purification Technology 2023, 304, 122320.[15] Korenč, M., Mencin, N., Puc, J., Skok, J., Nemec, K. Š., Celjar, A. M., Gagnon, P., Štrancar, A., Sekirnik, R. Cell and Gene Therapy Insights 2021, 7, 1207–1216.[16] Baiersdörfer, M., Boros, G., Muramatsu, H.,. Molecular Therapy - Nucleic Acids 2019, 15, 26–35.